医疗器械监管法律法规如何与时俱进?
 摘要:
中国的医疗器械监管法律体系是一个以《医疗器械监督管理条例》为核心,由多部法律、行政法规、部门规章、技术指导原则和标准构成的层级分明、覆盖全面的体系,其核心目标是保障医疗器械的安全、...
摘要:
中国的医疗器械监管法律体系是一个以《医疗器械监督管理条例》为核心,由多部法律、行政法规、部门规章、技术指导原则和标准构成的层级分明、覆盖全面的体系,其核心目标是保障医疗器械的安全、... 中国的医疗器械监管法律体系是一个以《医疗器械监督管理条例》为核心,由多部法律、行政法规、部门规章、技术指导原则和标准构成的层级分明、覆盖全面的体系,其核心目标是保障医疗器械的安全、有效,保障人体健康和生命安全。
(图片来源网络,侵删)
以下将从法律框架、核心内容、监管机构、产品生命周期监管四个维度进行详细解读。
法律框架体系
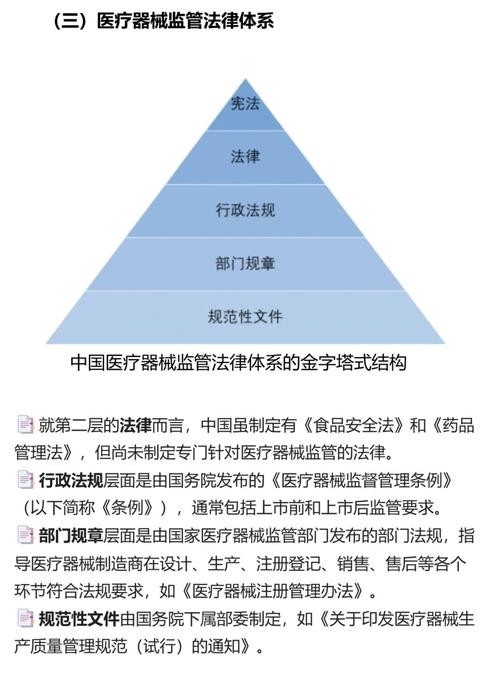
中国的医疗器械监管法律体系可以看作一个金字塔结构,从上到下依次为:
-
法律层面
- 《中华人民共和国药品管理法》: 虽然主要针对药品,但其总则部分也适用于医疗器械,确立了药品和医疗器械监管的基本原则,如风险管理、全程管控等。
-
行政法规层面
 (图片来源网络,侵删)
(图片来源网络,侵删)- 《医疗器械监督管理条例》: 这是医疗器械领域的“宪法”,由国务院发布,是所有医疗器械法规、规章和规范性文件的上位法,它规定了监管的基本原则、监管部门的职责、医疗器械的分类、研制、生产、经营、使用、监督管理的全过程以及法律责任,2025年新修订的条例是当前监管的基础。
-
部门规章层面
- 由国家药品监督管理局(NMPA)发布,是对《条例》的具体化和细化,是日常监管操作的主要依据。
- 《医疗器械注册与备案管理办法》: 规定了医疗器械产品如何进行注册(高风险)和备案(低风险)。
- 《医疗器械生产监督管理办法》: 规定了医疗器械生产企业的开办条件、生产质量管理规范以及日常监管要求。
- 《医疗器械经营监督管理办法》: 规定了医疗器械经营企业的开办条件、经营质量管理规范以及日常监管要求。
- 《医疗器械使用质量监督管理办法》: 规定了医疗机构如何采购、验收、储存、使用和维护医疗器械。
- 《医疗器械标识系统规则》: 推行医疗器械唯一标识(UDI),实现产品全生命周期追溯。
-
技术指导原则与标准
- 技术指导原则: NMPA发布的各类技术审查指南、临床评价指南、说明书和标签撰写指南等,为企业注册申报和合规生产提供具体操作指引。
- 国家标准和行业标准: 包括强制性标准和推荐性标准,规定了医疗器械的技术要求、检测方法、风险管理等,GB 9706系列是医用电气设备安全的核心标准。
核心内容与原则
《医疗器械监督管理条例》确立了几大核心原则和制度:
医疗器械分类管理
这是监管体系的基础,根据风险程度,将医疗器械分为三类:

(图片来源网络,侵删)
- 第三类(高风险): 植入人体,支持维持生命,对人体具有潜在危险,对其安全性、有效性必须严格控制的医疗器械。
- 心脏起搏器、人工关节、植入式神经刺激器、无菌医疗器械(如一次性注射器)等。
- 第二类(中风险): 具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
- 医用成像设备(如B超机)、体外诊断试剂、血压计、医用缝合线等。
- 第一类(低风险): 风险程度较低,实行常规管理可以保证其安全、有效的医疗器械。
- 医用纱布、口罩、手术衣、创可贴等。
监管力度:第三类 > 第二类 > 第一类。
产品准入:注册与备案
- 备案: 第一类医疗器械和部分第二类医疗器械(如部分医用成像器械、部分物理治疗器械等)实行备案管理,企业向省级药监局提交备案资料即可,流程相对简单。
- 注册: 第二类、第三类医疗器械必须向国家药品监督管理局(NMPA)或其授权的省级药监局申请注册,注册流程复杂,需要提交包括产品技术要求、检验报告、临床评价资料、说明书、标签样稿等在内的全套技术文件,并经过严格的审评审批。
生产质量管理规范
- 《医疗器械生产质量管理规范》: 类似于药品的GMP,是医疗器械生产企业的基本准则,它要求企业建立从原材料采购、生产过程控制、成品检验、储存运输到售后追溯的全过程质量管理体系。
- 体系核查: NMPA在注册审批和日常监管中,会对企业的质量管理体系进行现场核查,确保其持续合规。
经营质量管理规范
- 《医疗器械经营质量管理规范》: 针对医疗器械经营企业(批发商、零售商),要求其在采购、验收、储存、销售、运输等环节建立质量管理体系,保证产品在流通过程中的质量安全。
- 经营许可/备案: 第三类医疗器械经营企业需要取得《医疗器械经营许可证》;第二类医疗器械经营企业实行备案管理;第一类医疗器械无需许可和备案。
全生命周期监管与追溯
- 唯一标识: 强制推行UDI系统,为每一件医疗器械赋予“身份证”,实现从生产、流通到使用的全程可追溯。
- 不良事件监测与再评价: 建立医疗器械不良事件监测制度,要求生产经营企业和医疗机构主动报告不良事件,对已上市的高风险医疗器械,开展持续的安全性、有效性再评价。
- 召回制度: 当医疗器械存在缺陷,可能对人体造成伤害时,生产企业必须主动召回,监管部门可以责令其召回。
主要监管机构
- 国家药品监督管理局: 国务院直属机构,是医疗器械监管的最高机构,负责全国医疗器械的监督管理工作,如制定法规、标准,审批第三类医疗器械注册等。
- 省级药品监督管理局: 负责本行政区域内的医疗器械监督管理工作,如审批第二类医疗器械注册、第一类和部分第二类医疗器械备案、医疗器械生产/经营许可/备案等。
- 市场监督管理部门: 负责对医疗器械的价格、广告、反垄断等进行监管,并与药品监管部门协同执法。
- 卫生健康委员会: 负责对医疗机构的医疗器械使用行为进行监督管理,确保其在临床中合理、安全使用。
产品全生命周期监管流程
一个典型的医疗器械产品从上市到退市的全过程监管如下:
| 阶段 | 核心法规/要求 | |
|---|---|---|
| 研发与临床试验 | 《医疗器械临床试验质量管理规范》 | 企业需进行产品研发,并通过临床试验(豁免除外)来证明产品的安全性和有效性。 |
| 注册/备案 | 《医疗器械注册与备案管理办法》 | 根据产品分类,向NMPA或省级药监局提交注册申请或备案,获得注册证/备案凭证。 |
| 生产许可 | 《医疗器械生产监督管理办法》 | 建立符合《医疗器械生产质量管理规范》的质量管理体系,并取得《医疗器械生产许可证》。 |
| 经营许可/备案 | 《医疗器械经营监督管理办法》 | 经营企业需取得经营许可证或完成备案,并遵守经营质量管理规范。 |
| 上市后监管 | 《医疗器械使用质量监督管理办法》等 | 医疗机构需建立采购、验收、使用、维护制度,监管部门进行飞行检查、日常监督检查。 |
| 不良事件监测与召回 | 《医疗器械召回管理办法》 | 企业和医疗机构主动报告不良事件,监管部门评估后,必要时启动产品召回程序。 |
| 再评价 | 《医疗器械监督管理条例》 | 对高风险或问题产品,NMPA可要求企业开展再评价,以持续评估其风险和收益。 |
中国的医疗器械监管法律法规体系正在不断完善,其特点是:
- 风险导向: 以产品风险等级为基础,实施差异化管理。
- 全程管控: 覆盖从研发、生产、经营到使用、召回的全生命周期。
- 国际接轨: 积极借鉴和采纳国际先进的监管原则和技术标准,如ISO 13485质量管理体系、人用药品注册技术要求国际协调会指南等。
对于医疗器械行业从业者而言,深刻理解并严格遵守这些法律法规,是企业合规经营和持续发展的基石。
文章版权及转载声明
作者:99ANYc3cd6本文地址:https://nbhssh.com/post/6045.html发布于 02-20
文章转载或复制请以超链接形式并注明出处宁波恒顺财经知识网